
医薬品の業態許可

1.医薬品とは
日本における医薬品等の取扱いについては、「医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律(医薬品・医療機器法、従前の薬事法)」によって規定されています。この法律における医薬品とは、
- 日本薬局方に収められている物
- 人又は動物の疾病の診断、治療又は予防に使用されることが目的とされている物であって、機械器具等でないもの(医薬部外品及び再生医療等製品を除く)
- 人又は動物の身体の構造又は機能に影響を及ぼすことが目的とされている物であって、機械器具等ではないもの(医薬部外品、化粧品及び再生医療等製品を除く)と定義されています。
医薬品は病気の診断、治療又は予防に使用されます。特に治療に用いられる医薬品には人体に対して強い作用を示す物もあるため、正しい使い方をしなければ体に悪い影響を与えることがあります。こうした医薬品を取扱うためには専門の知識を有する責任者や必要な設備、対応できる人的組織などが必要とされています。こうした医薬品の製造、輸入や販売などの業態を行う者に対して許可されるものを業態許可と言います。
2.製造販売業について
業態許可のなかでも大変重要な許可が製造販売業の許可で、医薬品の市場での責任をより明確にするために平成14年(2002年)の旧薬事法改正で新たに導入された許可制度です。
製造販売とは、医薬品・医療機器法では「製造(他に委託して製造した場合も含む)又は輸入した医薬品を日本国内に販売等を行い提供すること」と定義されており、この製造販売を行うためには「医薬品製造販売業許可(製販業許可)」を取得しなければなりません。
製販業許可を取得した者(以下「製販業許可者」)は、販売によって市場にある医薬品に対して最終的な責任を負う者であり、製品の副作用情報、クレーム情報、事故情報等を国内外から積極的に収集し、市販後の製品について安全管理を行うとともに、万が一製品に何らかの問題があると判断した場合には、必要に応じて適切な措置を行うなど非常に大きな責任を有することになります。また、併せて医薬品の製造においても、適正な品質管理のもとで製品が製造されいるかを管理監督する義務も有しています。
このように製販業許可者は、市場にある当該医薬品に対して最も重い責任を負う者であり、製品の品質管理及び市販後の医薬品についての安全管理を行うことが出来る能力が求められます。
なお、製販業許可で取り扱う医薬品は、品目ごとにその製造販売については厚生労働大臣の承認を受けなければなりません。この承認を受けた医薬品は製販業許可のみでは製造することはできません。
医薬品を製造するためには「医薬品製造業許可(製造業許可)」という医薬品製造に特化した許可を別に取得しなければなりません。製販業許可者が自ら製造業許可を取得し医薬品を製造する場合もありますが、製造業許可のみを取得した医薬品製造に特化した業者に製造委託(一部委託並びに全面委託)を行う場合もあります。製造委託を行う場合は、製販業許可者の厳格な管理監督の元に製造することになります。これは承認を得た医薬品の市場における責任は製販業許可者にあるためであり、そのため製造業許可のみでは製品を市場に出荷することが出来ない仕組みになっています。
また、製販業許可者が出荷した医薬品については、薬局開設者または医薬品の販売業の許可がなければ販売、授与又は販売もしくは授与の目的で貯蔵し、陳列することは出来ません。
3.製販業許可について
製販業許可を取得するためには、以下の要件を満たす必要があります。
- 品質管理の方法が、医薬品等の品質管理の基準に関する省令(GQP省令)に適合している。
- 製造販売後安全管理の方法が、医薬品等の製造販売後安全管理の基準に関する省令(GVP省令)に適合している。
- 申請者(法人の場合はその業務を行う役員等)が欠格条項に該当しない。
- 総括製造販売責任者を設置すること。
上記の1については、品質管理業務について責任を有する者として、「品質保証責任者」を設置し、市場への出荷の管理、製造業者等の取決め、適正な製造管理及び品質管理の確保、品質等に関する情報及び品質不良等の措置などを行うことになります。
また2については、製造販売後安全管理業務について責任を持って行うため、「安全管理責任者」を設置し、医療関係者・学会報告、文献・行政、海外当局からの情報などの安全管理情報の収集・検討や添付文書の改訂などの安全性確保措置の立案と実施などが適切に行えることが求められています。
また、3の欠格条項とは、以前に医薬品の業態許可が取り消されたり、申請者が禁固以上の刑に処されたり、麻薬等の中毒者であるなど、医薬品の製造販売に関する責務が行えないような場合が該当しています。
医薬品の品質と安全性に関する情報は個別の案件ではなく、それぞれ関連して検討を行い総合的に第三者的な視点での判断が求められています。そうした判断を行う者として4の総括製造販売責任者を設置しなければなりません。
総括製造販売責任者は、品質保証責任者、安全管理責任者を監督し、これら責任者からの報告に基づき措置の決定、決定した措置の実施を適切な実施権限者に指示し、必要があると認める場合には、製造販売業者つまり製販業許可を有する企業等の経営陣や経営者に対して意見を述べる必要があります。
この総括製造販売責任者、品質保証責任者、安全管理責任者は、製販業許可の三役制度と称されています。
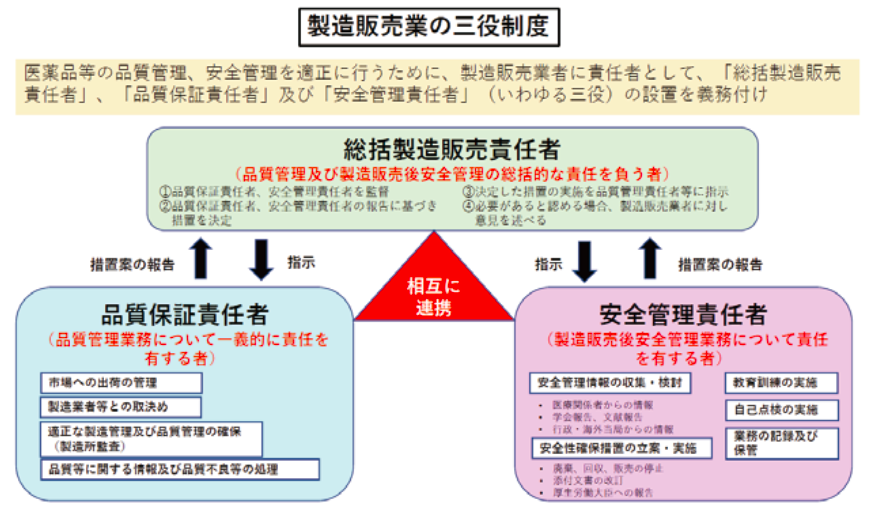
品質保証責任者の要件はGQP省令では、品質保証部門の責任者であり、また品質管理業務等に3年以上従事した者であり、当該業務を適正かつ円滑に遂行しうる能力を有するものと規定されています。
また安全管理責任者の要件もGVP省令において、安全管理統括部門の責任者であり、当該業務等に3年以上従事した者であり、当該業務を適正かつ円滑に遂行しうる能力を有するものであると規定されています。
一方、総括製造販売責任者の資格は、処方せん医薬品を扱う場合には、薬剤師であることのみが規定されており、業務経験については現状では何も規定されていません。
4.製造販売承認
製販業許可で取り扱う医薬品は、品目ごとにその製造販売について厚生労働大臣の承認を受けることが医薬品・医療機器法で規定されています。ここで記載されている承認とは、製造販売される医薬品の品質、有効性及び安全性等の観点から、医薬品として適当であるか否かが判断されます。承認を取得するためには製造販売される品目毎に承認申請を行いますが、その承認申請には当該医薬品の一貫した品質を保証するため、その医薬品の詳細な製造方法、品質が担保されていることを確認する品質規格ならびに規格試験法などを規定する必要があります。
承認申請書で規定し、厚生労働大臣が承認した製造方法通りに製造しなければなりませんが、製造物である医薬品では、場合によって温度、湿度などの外的環境の変化による製造方法の変更や、より高品質な医薬品を製造する目的での製造方法の改良など、変更が生じる必要があります。そうした変更についても製造方法の細かい内容は厚生労働大臣により承認されていますので、必ず変更の薬事的な手続きを行わなければなりません。製販業許可者の中には多くの品目を製造販売しているケースもあり、こうした製造方法の変更と承認事項の薬事的な手続きにギャップ生じるようなことも場合によっては起こることもありますが、このようなことが起こらないよう製販業許可の三役体制による確実な相互の連携と密なコミュニケーション並びに厳格な責任体制の構築などが求められることは言うまでもありません。
JGAニュースNo.108(2017年4月号)


