
「承認書と製造実態との自主点検について」
2024年4月5日付で「後発医薬品の製造販売承認書と製造方法及び製造方法及び試験方法の実態の整合性に係る点検の実施について」の通知が発出されました。この通知は厚生労働省の医薬局 医薬品審査管理課長、監視指導・麻薬対策課長、医政局 医薬産業振興・医療情報企画課長など医療用医薬品に関して主な行政上の管理・監督を行う部署の三課長により発出されたものですが、その内容は薬価収載されているすべての後発医薬品(バイオシミラー、バイオセイムを除く)の承認を有する製造販売業者に対して、本年10月末までに製造販売承認書(以後「承認書」と称します。)と製造実態の整合性に関する点検を行うことを求めるものです。
医療用医薬品は、承認書に記載された製造方法で製造され、また記載された試験方法に基づき、その規格に合格したものが、定められた用法・用量に従って使用されることにより、はじめて承認された効能・効果が期待できます。これら承認書に記載されている内容は、規制当局による厳格・適切な審査を受けたものですので、承認書に従った製造や試験が行われることにより製品品質が保たれることになります。
しかしながら、製造物として製造される医薬品については、科学技術の進歩や新しい研究開発により、更に効率的でより高品質な医薬品を製造することが検討されるなど、日々革新的な進歩を遂げているケースもあります。承認書に記載されている製造方法や試験方法は、その承認を得た時点では最新の技術が取り入れられおり、これらの新しい技術による製造や試験法は承認書に記載されている内容から科学的に読み取れられるものもありますが、承認書に記載されている内容については、必要に応じて記載内容の見直しや変更等が必要になる場合があります(承認書の変更に係る薬事対応については、JGAニュース 2022年1月号_No.165「一変申請」をご参照下さい)。
ただ、科学的に十分な評価を行わずに承認書に記載されている必要なステップや手順を省略するような製造行為、試験の実施や、根拠に乏しい経験則に基づく変更が行われた製造行為、試験の実施などは、承認書に記載された内容に基づいていない製造行為になる場合があります。また、新しい技術による製造方法や試験方法などについても必要に応じて適切に承認書の記載を変更する必要があるケースもありますが、手続きの問題で承認書の変更が行われていない場合もありますので、医薬品の製造販売業者は常に承認書の内容については確認を行うことに加え必要な薬事対応を行うことが求められています。
まず今回の点検に至った経緯について説明させていただきます。2015年に血液製剤及びワクチン製剤の製造を行う企業が、20年以上にわたり、虚偽の記録を作成するなど不正な隠蔽を図るなどにより、承認書とは異なる製造方法で製造を行っていたことが発覚しました。医療用医薬品は国民の健康維持向上に必要な重要製品であることから、こうした事案を踏まえて、厚生労働省は、2016年に全ての医療用医薬品の製造販売業者に対して、承認書と製造実態の整合性に係る点検の実施を指示しました。これは当時、医療用医薬品の一斉点検と称されました。この点検では全医薬品32,466品目について、646社の医薬品製造販売業者による点検が実施され、2016年6月1日にその点検結果が厚生労働省より公表されています。それ以降も承認書と異なる製造を行うという事案が散見されるケースがあり、2020年12月以降に発覚しました後発医薬品企業による医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律(以後「薬機法」と称します。)に違反する行為などにより失った後発医薬品の信頼回復を目的として、2021年に日本ジェネリック製薬協会(以後「GE薬協」と称します。)は、2016年の一斉点検から5年を経過したこともあり、承認書と製造実態との整合性を統一した手法で確認を行う自主点検を実施致しました。その後も残念ながら後発医薬品企業による製造管理・品質管理上の不備による薬機法に違反するような事例が頻発し、それに伴う後発医薬品の安定供給に対する改善がなされていない状況が不本意ながら継続しています。今回の三課長通知による承認書の自主点検は、この事態を打開するための実効性のある実態調査を今一度行うことにより、国民の皆さまの後発医薬品への信頼を回復するための最後の機会が与えられたという位置づけとされています。
そのためにも、後発医薬品企業においては、当該点検を適切に、真摯に対応し、医療に貢献する社会インフラの一部としての矜持を堅持する義務があると考えています。今回の通知では、自己点検の途中経過を各社の自社ホームページ上で公表するとともに、医薬品業界団体を統括する日本製薬団体連合会(以後「日薬連」と称します。)にも途中経過の報告を行うことが求められています。更に、自主点検が終了した時点で、その点検結果を規制当局並びに日薬連にも報告する必要があります。
今回の自主点検は今までに行われた点検とは異なる特徴がありますので、この点についてお示し致します。
2016年の一斉点検は、各社各様の手法で点検が実施されました。また、2021年のGE薬協の自主点検は、協会会員の全社がGE薬協で設定した統一的な点検手法に基づく点検を実施しましたが、GE薬協会員外の後発医薬品の製造販売業者まで全て実施されているという状況ではありませんでした。そのため今回は日薬連の主導により、薬価基準に収載された全ての後発医薬品を製造販売する製造販売業者によって、初めて同じ手法・基準による点検が行われることになります。
まず、承認書と製造及び試験に関連する作業手順書や製造・試験に関する全ての記録書を一文字一句(Line by Line) で比較を行い、すべての相違を洗い出した後に、製造管理および品質管理の基準「Good Manufacturing Practice」(以後、「GMP」と称します。)で許容されている相違なのか、許容されていない相違(この場合は承認書への薬事対応が必要な齟齬と考えられます)なのかを判断致します。
書面のみの調査だけではなく、今回は実際の製造現場で作業する従業員の方々へのヒアリングも実施し、現在行われている作業は承認書をベースとした作業手順書に従っているものか、或いは前任者や先輩従業員等からの口頭伝承や不文律等による作業なのかを確認するなど、書面確認だけでは検知できない承認書と実態の相違の有無についても点検を行うこととしています。
規格及び試験方法の点検においては、2023年6月21日付の薬生薬審発0621第4号・薬制監麻発第5号で発出された代用法通知(代用法については、JGAニュース2023年10月号_No.186「代用法について」をご参照下さい。)、および代用法通知に関する質疑応答集(Q&A)(2024年6月24日付審査管理課・監麻課発事務連絡)も参考として、その実態を点検することとなっています。
今回の自主点検の着手にあたり、日薬連が通知で提示した統一的な点検手法による製造方法欄の点検は、GE薬協が2021年に実施した点検手法が採用されましたが、規格及び試験方法欄・別紙規格欄の点検については、GE薬協の点検手法と同じ考え方を踏襲しつつ、新たにデータインティグリティ(Data Integrity:データの完全性と正確性が客観的に担保されていること)や昨今の不正事例もチェックできるよう配慮された方法を採用しています。規格及び試験方法欄等の点検には、GE薬協の薬制委員会で原案が検討された点検フローがベースとなっており、外部専門家による監修を行っていただいた後に、厚生労働省の関係部局の確認後、日薬連にて最終化されたものです。2024年6月25日には、今回の承認書自主点検に関する日薬連主催による説明会が開催され、承認書自主点検通知を発出した厚生労働省の医薬局 医薬品審査管理課、監視指導・麻薬対策課、医政局 医薬産業振興・医療情報企画課の各課担当者による説明に加え、日薬連の担当プロジェクトのリーダーによる説明が行われています。その説明会において、点検対象品目を有する全ての製造販売業者に対して、本点検フローが提示され、点検フローを用いた点検方法に関する説明も行われています。なお、当該点検フローは2024年6月25日付で発出された「後発医薬品の製造販売承認書と製造方法及び試験方法の実態の整合性に係る代用試験法を使用している場合の試験方法の自主点検実施手順について」(日薬連発第431号)の別添資料として公開されています。日薬連では、今後自主点検によって顕在化すると考えられる点検上の疑問点(相違なのか齟齬なのかの判断等)や齟齬の場合の薬事対応の考え方も含め、原則すべての情報を自主点検に参加した製造販売業者と共有していく方針が示されました。
今回の日薬連主催で開催された承認書自主点検の説明会では、代用法通知のQ&A通知の発出を踏まえた規格及び試験方法欄、別紙規格欄の点検方法に関する点検フローの説明が行われていますが、留意すべき事項を含め、その概略について次のように解説致します。
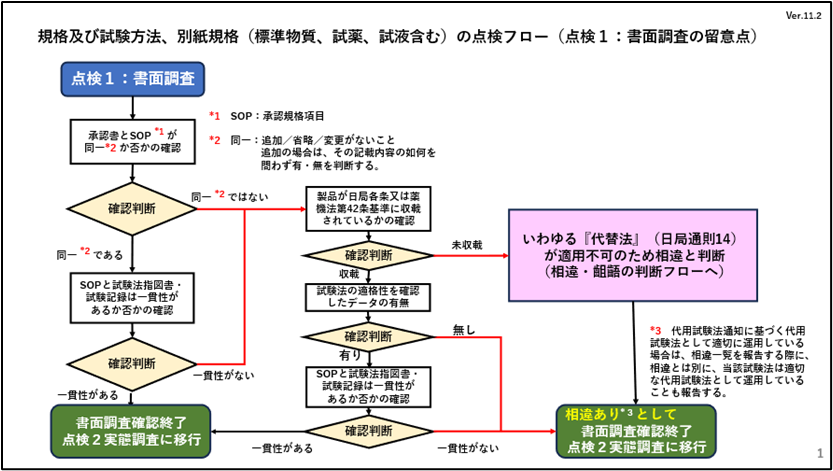
「点検1 書面調査」
承認書とSOP(手順書)の書面による同一性のチェックにおいて、日本薬局方の医薬品各条に収載されている品目または薬機法の第42条の医薬品等の基準に収載されている品目か否かで、その同一性の許容範囲が異なることになります。これらに収載されている品目については、日本薬局方の通則141)の適用を受け、規定の方法以上の真度及び精度がある場合には規定する試験法に代わる方法で試験を実施することができることが“代用法通知”に示されおり、代替試験法として認められます。そのため代替試験法の場合は承認書に規定されている試験法(承認法)と異なっていても相違には該当しません。しかしながらそれ以外の品目については、試験法の少しの相違であっても、承認法には該当せずに別法となるため、承認書の変更などの薬事対応が必要になります。
ただし、日本薬局方の通則14が適用される代替試験法に該当する場合においても、その代替試験法を適用することを検証したバリデーションデータの存在や、SOPに代替試験法を使用する旨を明記することが必須条件となります。
今回、点検フローに記載していますSOPと試験指図書・試験記録の一貫性のチェックにおいては、同一という用語は用いず、一貫性という用語を用いています。これは、同一であれば、承認書とSOP等が完全に一文字一句一致しなければ相違に該当するためであり、同様の内容であることを示す必要な記録等が残せるフォーマットを使用していれば、一貫性があり問題ないことが判断できると考えました。
1)日本薬局方の通則14:日本薬局方に規定する試験法に代わる方法で、それが規定の方法以上の真度及び精度がある場合は、その方法を用いることができる。ただし、その結果について疑いのある場合は、規定の方法で最終の判定を行う。
1.点検フロー(点検1:書面調査)
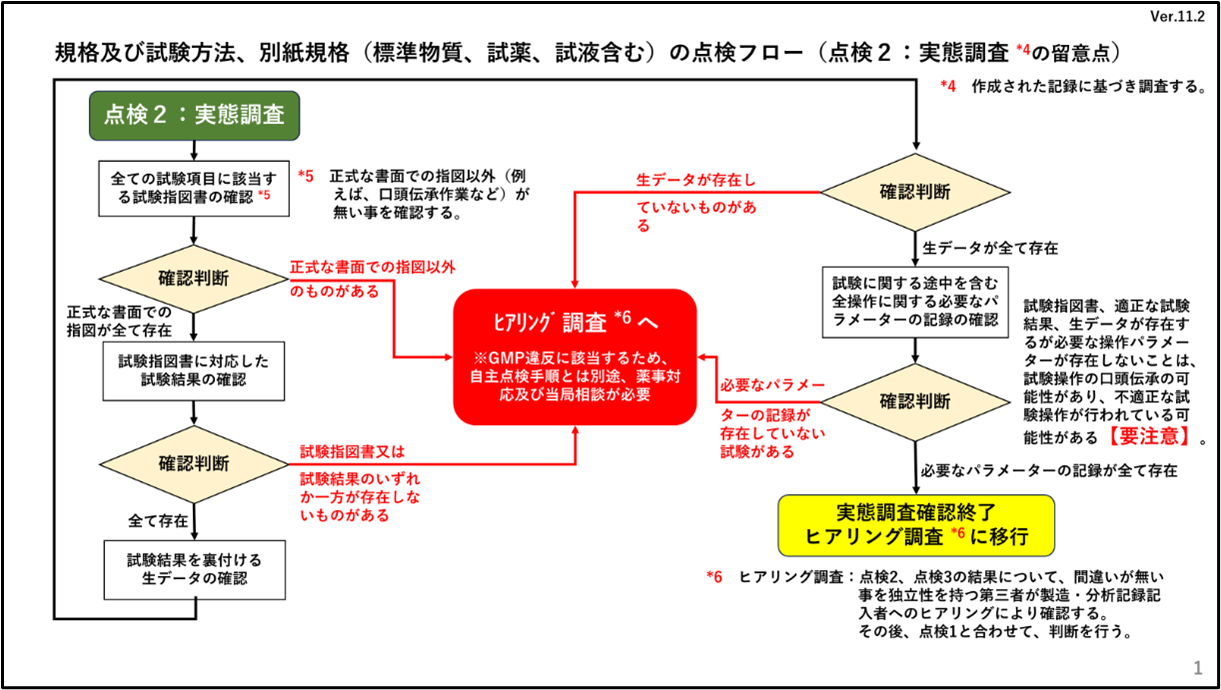
「点検2 実態調査」
点検1の結果において、承認書とSOP等に「相違あり」或いは「相違なし」の結果に拘わらず、書面の調査だけでは点検漏れとなる可能性があるため「点検2 実態調査」の点検を行うこととしています。ここでは、昨今発生している品質に関する不正事案を参考にして、承認書通りに試験は実施されているが、実際には適合となるように試験データの改ざんが行われていないかという点についてチェックを行います。実際に試験実施により作成された試験記録に基づき、正式な指示の下、指示された通りに試験に係る諸作業が実施されたかどうかの確認を行います。このチェックで、試験記録として明らかにデータが不足するような事例がある場合は、GMPで求められていますデータインティグリティ(Data Integrity)が担保できない事態となり、製造・品質管理に係るガバナンス上の問題が懸念されることになります。したがって点検2に記載されています次の4つの確認項目の資料は、適切に試験が行われていれば、必ず該当する資料・記録が存在しているものであり、これらについては、すべて確認を実施する必要があります。
①全ての試験項目に該当する正式な書面での試験指図書
②試験指図書に対応した試験結果のデータ等の記録
③当該試験結果を裏付ける試験実施の生データの記録及び資料
④試験を実施する際の全試験操作に必要なパラメータの記録
このチェックの際には、例えば試験条件なども含めた試験に関する全ての記録を確認します。これを確認終了後、作業員に対するヒアリング調査の実施に移行します。
2.点検フロー(点検2:実態調査)
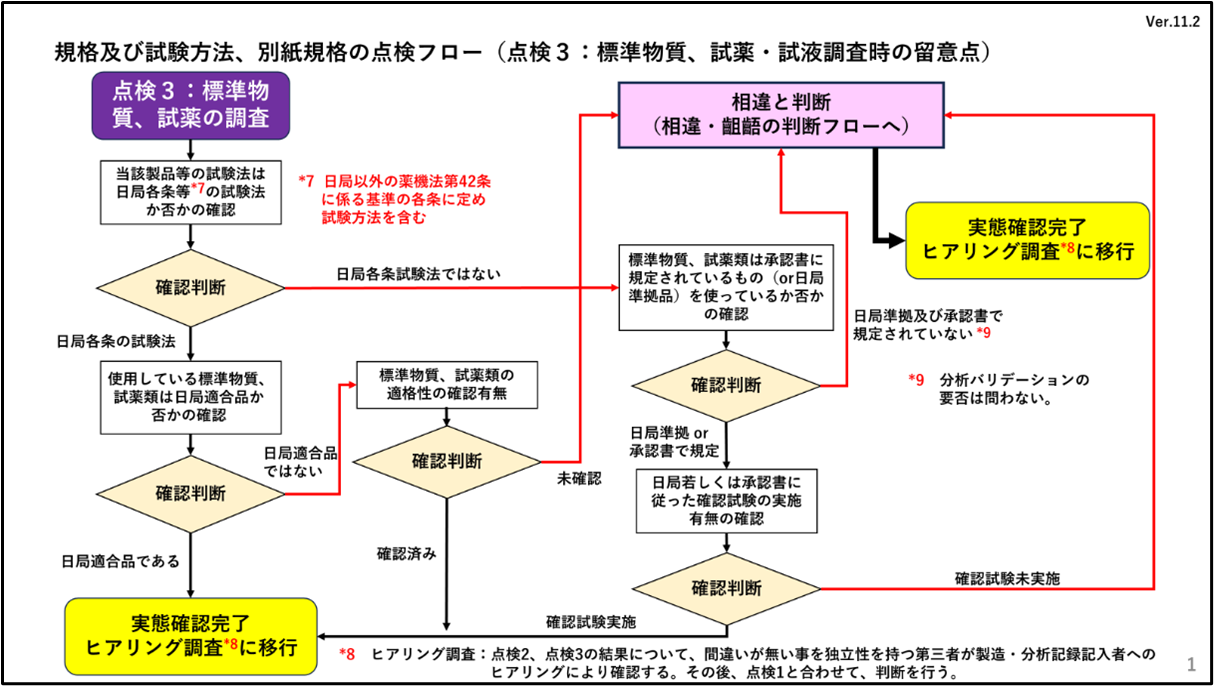
「点検3 標準物質、試薬・試液調査」
この点検は、規格及び試験方法に特徴的な調査になります。本来、試験に使用する試薬類については、その品質を確認するためにすべて規格が設定されています。承認書の規格及び試験方法欄の備考には、通常、「本規格及び試験方法は、別に規定するもののほか、日本薬局方通則、製剤総則及び一般試験法を準用する。」と記載されており、特に規定されていない場合は、日本薬局方の試薬・試液に規定されている規格適合品を使用しなければなりません。一方、“代用法通知”およびその“Q&A”に従えば、日本薬局方の各条や薬機法第42条の基準に収載された品目の試験法以外では、グレード違い(同一の試薬でも特級、一級など異なるものがある)の試薬の継続使用は別法扱いとなり認められないことから、この「標準物質、試薬・試液」の点検も必ず実施されることを意図して、点検フローを作成しています。
3.点検フロー(点検3:標準物質、試薬の調査)
「ヒアリング調査」
点検2,3が終了すればこのヒアリング調査に移行します。この点検作業においては、特に作業に係る詳細な手順は作成していませんが、直近の不正事案を鑑み、記録書等の書面による点検チェックだけでは判らない、作業員の故意によるもの、あるいは故意によらないもの全てについて、指示書・記録書には反映されない口頭伝承や不文律、慣習として実施されていた作業や実施すべき作業を実施してなかった事項の有無について、作業者である従業員に直接ヒアリングを行う点検になります。なお、ヒアリング対象者の心理的安全性を考慮(公益通報者保護制度の説明等)しつつ、少なくとも次の4項目を確認することになります。
①手順で定められた事項はすべて実施したか?
②手順に定められていない事項を実施してないか?
③口頭伝承や不文律として記録に残らない作業の実施がないか?
④他の担当者が不正を行っていることを見聞きしたことがないか?
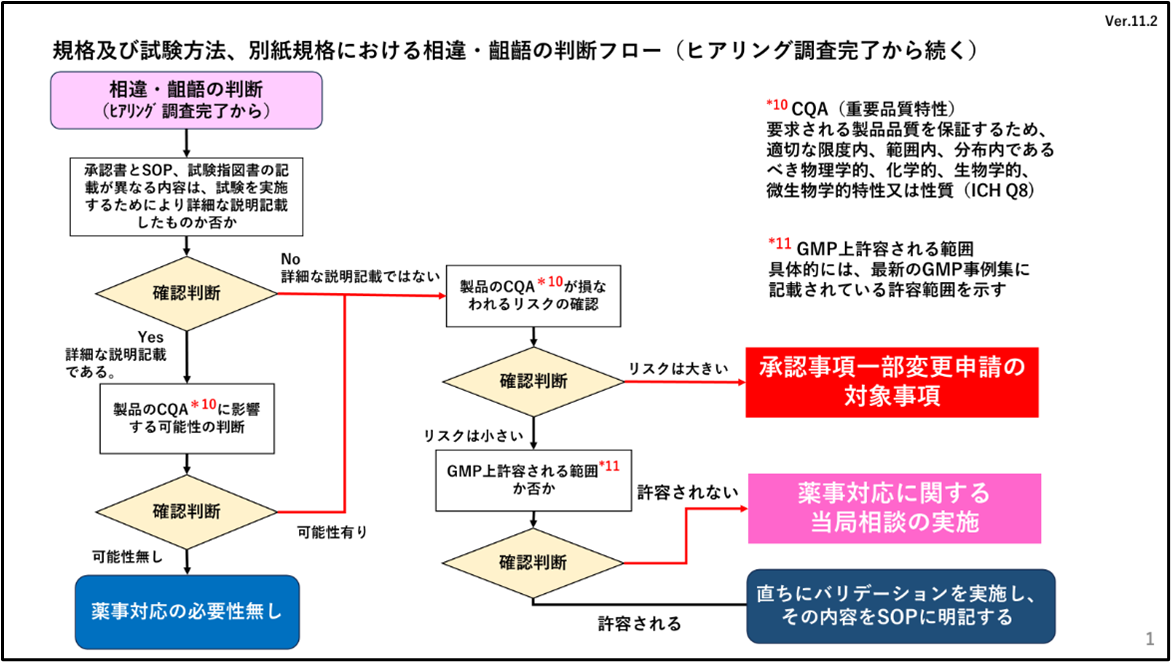
「相違・齟齬の判断」
この判断フローは、製造方法欄の点検時のものと同一の考え方です。ここで、CQA(Critical Quality Attribute:重要品質特性)が損なわれるリスクの大小についてはいろいろなケースがありますので、一般化が可能な疑問点については、日薬連が窓口になり随時質問を受付け、薬事対応の考え方については本自主点検を行う全社で共有していく方針です。日薬連が指定するフォーマットを用いて日薬連の対応窓口に連絡するスキームが設けられています。
4.点検フロー(相違・齟齬の判断)
最後に、GE薬協会員会社は、承認書の製造実態と製造方法欄の点検については、2021年のGE薬協自主点検により、今回の点検開始時点で会員各社が承認を有する後発医薬品については、書面調査・薬事対応が完了しており、以降新たに薬価収載された製品の点検、および今回の三課長通知で実施が求められている作業者へのヒアリング点検を行うことになります。また、規格及び試験方法欄についても、“代用法通知”が発出された時点から点検に関する準備を行っていた会員会社もありますが、今般“代用法通知”の“Q&A通知”も発出され、より明確になったその判断基準に基づき、三課長通知に従った確認作業を進めることになります。今回の点検に至った背景にも記載しましたが、今回の自主点検を、後発医薬品への信頼回復の最後のチャンスと捉え、期限を厳守した対応が求められていることを常に念頭に置いた点検が行われることが重要であると考えます。
【関連通知】
後発医薬品の製造販売承認書と製造方法及び試験方法の実態の整合性に係る点検の実施について【三課長通知】
(医政産情企発0405第2号、医薬薬審発0405第9号、医薬監麻発0405第2号)
令和6年4月5日
リンク
医療用医薬品の製造に当たり承認書の「別紙規格欄」及び「規格及び試験方法欄」に規定する試験方法に代用しうる試験方法を実施する場合の取扱いについて【代用法通知】
(薬生薬審発0621第5号、薬生監麻発0621第6号)
令和5年6月21日
リンク
「医薬品の製造に当たり承認書の「別紙規格欄」及び「規格及び試験方法欄」に規定する試験方法に代用しうる試験方法を実施する場合の取扱いについて」に関する質疑応答集(Q&A)について【代用法通知に関するQ&A】
(事務連絡)
令和6年6月24日
リンク
「後発医薬品の製造販売承認書と製造方法及び試験方法の実態の整合性に係る代用法試験法を使用している場合の試験方法の自主点検実施手順」について【別添点検フロー通知】
(日薬連発第431号)
2024年6月25日
リンク